INPS/INPS-3D: sequence- and structure-based prediction of ΔΔG upon variation
INPS/INPS-3D methods are described in the following articles:
- Castrense Savojardo, Piero Fariselli, Pier Luigi Martelli, Rita Casadio (2016)
INPS-MD: a web server to
predict stability of protein
variants from sequence and structure,
Bioinformatics, 32(16),2542–2544.
Bioinformatics PubMed - Piero Fariselli, Pier Luigi Martelli, Castrense Savojardo, Rita Casadio (2015)
INPS: predicting the impact of non-synonymous variations
on protein stability from sequence,
Bioinformatics, 31(17), 2816–2821.
Bioinformatics PubMed
Both INPS and INPS-3D adopt a Support Vector Regression (SVR) approach. In INPS, the SVR is trained on features extracted from the protein primary sequence, including:
- BLOSUM62 variant score (BL62)
- hydrophobicity (HYD) and molecular weights (MW) of native and variant residues
- Dayhoff mutability index (MI) of the native residue
- evolutionary information derived from multiple sequence alignments (EVI)
INPS-3D extends the feature set by also including information extracted from the protein 3D structure:
- Relative Solvent Accessibility (RSA)
- native/variant difference in overall pair-wise contact potential computed in a local structural environment (POT)
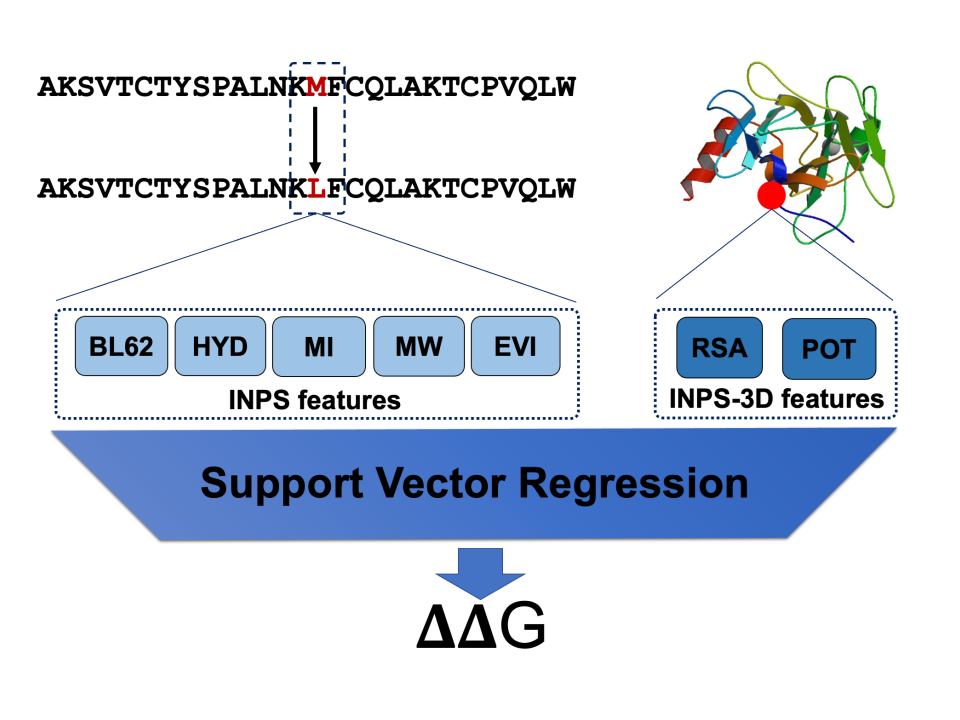
Figure 1. - INPS-SEQ and INPS-3D architectures
INPS and INPS-3D have been trained/tested using a dataset of 2648 variants occurring in 132 proteins (see Datasets). The dataset (S2648) was originally extracted from the ProTherm database and curated by the authors of the PoPMuSiC algorithm (Dehouck et al., 2009).
Five-fold cross-validation has been performed on the S2648 dataset. Cross-validation split has been computed at the protein level: all variations occurring in the same protein were collected into the same testing set avoiding any possible bias between training/testing data.
For sake of comparison with other approaches, an additional dataset has been used, comprising 42 variations within the DNA binding domain of the tumor suppressor protein p53, whose thermodynamic effects have previously been experimentally characterized (P53 dataset).
References
- Dehouck, Y. et al. (2011) PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics, 12, 151.